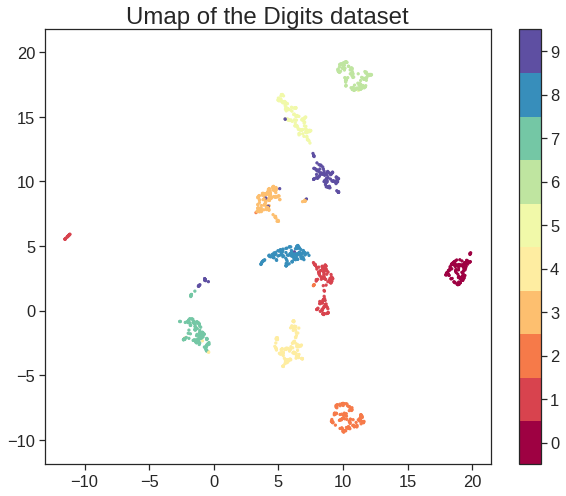
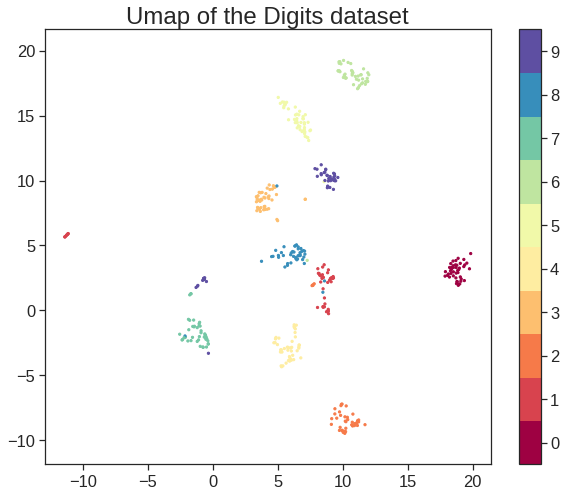
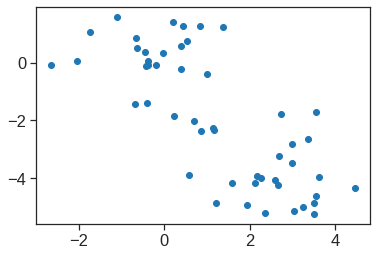
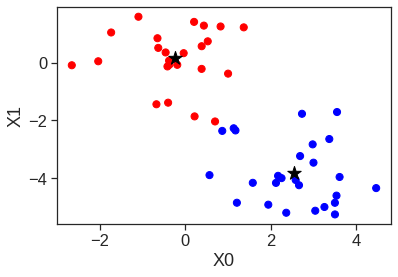
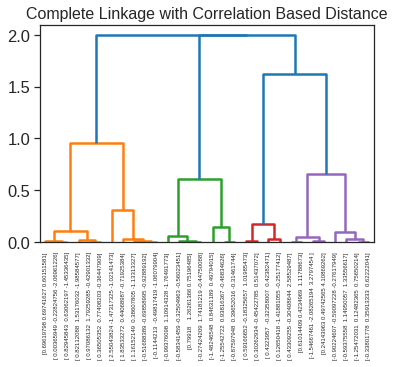
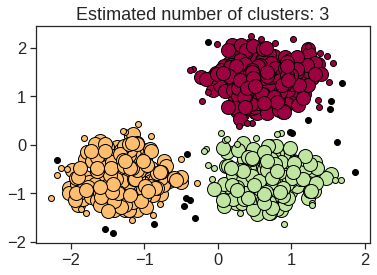
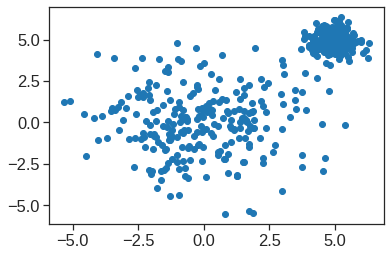
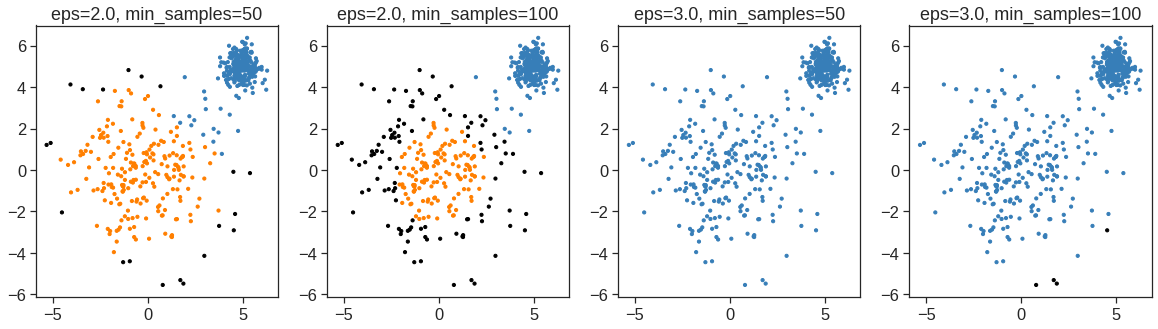
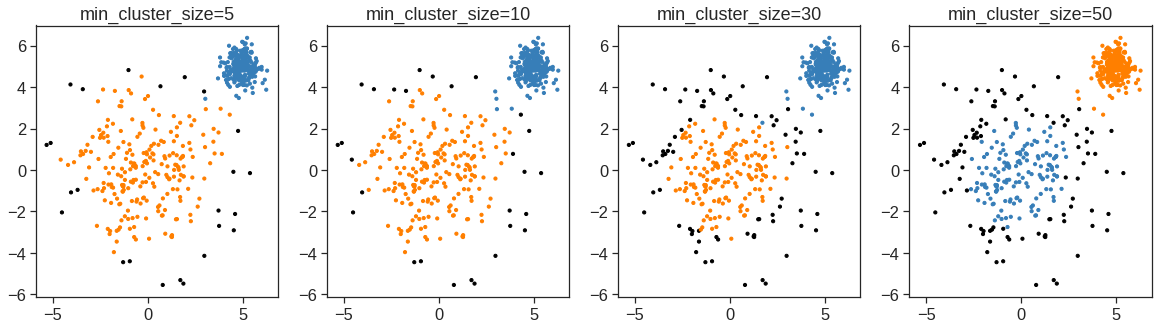
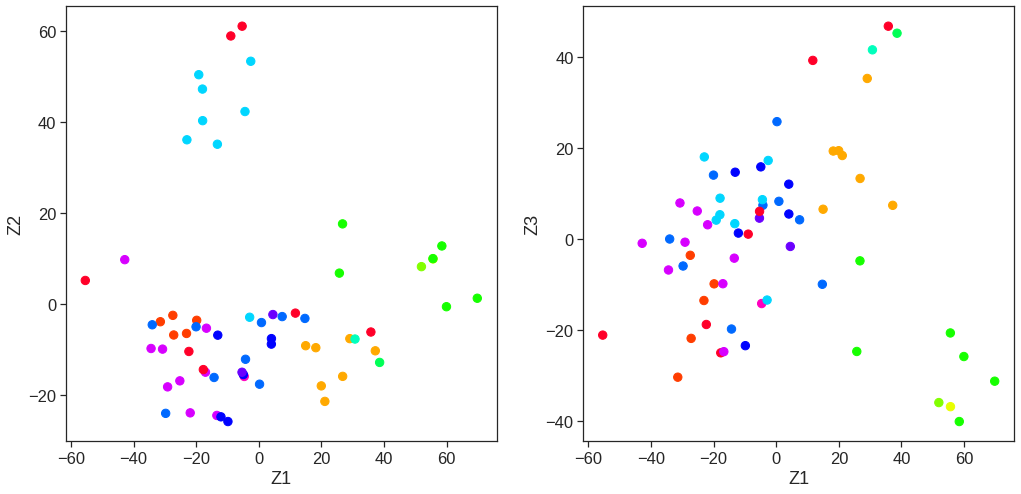
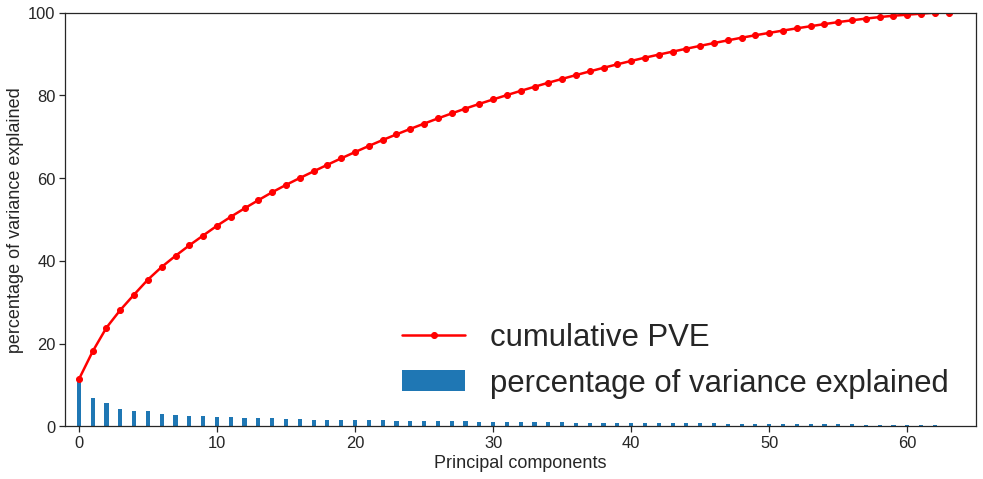
array([[ 9.85565885e-01, 1.13339238e+00, -4.44268788e-01,
1.56267145e-01],
[ 1.95013775e+00, 1.07321326e+00, 2.04000333e+00,
-4.38583440e-01],
[ 1.76316354e+00, -7.45956781e-01, 5.47808243e-02,
-8.34652924e-01],
[-1.41420290e-01, 1.11979678e+00, 1.14573692e-01,
-1.82810896e-01],
[ 2.52398013e+00, -1.54293399e+00, 5.98556799e-01,
-3.41996478e-01],
[ 1.51456286e+00, -9.87555085e-01, 1.09500699e+00,
1.46488703e-03],
[-1.35864746e+00, -1.08892789e+00, -6.43257569e-01,
-1.18469414e-01],
[ 4.77093091e-02, -3.25358925e-01, -7.18632942e-01,
-8.81977637e-01],
[ 3.01304227e+00, 3.92285133e-02, -5.76829492e-01,
-9.62847520e-02],
[ 1.63928304e+00, 1.27894240e+00, -3.42460080e-01,
1.07679681e+00],
[-9.12657146e-01, -1.57046001e+00, 5.07818939e-02,
9.02806864e-01],
[-1.63979985e+00, 2.10972917e-01, 2.59801342e-01,
-4.99104101e-01],
[ 1.37891072e+00, -6.81841189e-01, -6.77495641e-01,
-1.22021292e-01],
[-5.05461361e-01, -1.51562542e-01, 2.28054838e-01,
4.24665700e-01],
[-2.25364607e+00, -1.04054073e-01, 1.64564315e-01,
1.75559157e-02],
[-7.96881121e-01, -2.70164705e-01, 2.55533072e-02,
2.06496428e-01],
[-7.50859074e-01, 9.58440286e-01, -2.83694170e-02,
6.70556671e-01],
[ 1.56481798e+00, 8.71054655e-01, -7.83480358e-01,
4.54728038e-01],
[-2.39682949e+00, 3.76391576e-01, -6.56823864e-02,
-3.30459817e-01],
[ 1.76336939e+00, 4.27655192e-01, -1.57250127e-01,
-5.59069521e-01],
[-4.86166287e-01, -1.47449650e+00, -6.09497476e-01,
-1.79598963e-01],
[ 2.10844115e+00, -1.55396819e-01, 3.84868584e-01,
1.02372019e-01],
[-1.69268181e+00, -6.32261251e-01, 1.53070434e-01,
6.73168850e-02],
[ 9.96494459e-01, 2.39379599e+00, -7.40808395e-01,
2.15508013e-01],
[ 6.96787329e-01, -2.63354790e-01, 3.77443827e-01,
2.25824461e-01],
[-1.18545191e+00, 5.36874372e-01, 2.46889321e-01,
1.23742227e-01],
[-1.26563654e+00, -1.93953730e-01, 1.75573906e-01,
1.58928878e-02],
[ 2.87439454e+00, -7.75600196e-01, 1.16338049e+00,
3.14515476e-01],
[-2.38391541e+00, -1.80822897e-02, 3.68553932e-02,
-3.31373376e-02],
[ 1.81566110e-01, -1.44950571e+00, -7.64453551e-01,
2.43382700e-01],
[ 1.98002375e+00, 1.42848780e-01, 1.83692180e-01,
-3.39533597e-01],
[ 1.68257738e+00, -8.23184142e-01, -6.43075093e-01,
-1.34843689e-02],
[ 1.12337861e+00, 2.22800338e+00, -8.63571788e-01,
-9.54381667e-01],
[-2.99222562e+00, 5.99118824e-01, 3.01277285e-01,
-2.53987327e-01],
[-2.25965422e-01, -7.42238237e-01, -3.11391215e-02,
4.73915911e-01],
[-3.11782855e-01, -2.87854206e-01, -1.53097922e-02,
1.03323208e-02],
[ 5.91220768e-02, -5.41411454e-01, 9.39832977e-01,
-2.37780688e-01],
[-8.88415824e-01, -5.71100352e-01, -4.00628706e-01,
3.59061124e-01],
[-8.63772064e-01, -1.49197842e+00, -1.36994570e+00,
-6.13569430e-01],
[ 1.32072380e+00, 1.93340466e+00, -3.00537790e-01,
-1.31466685e-01],
[-1.98777484e+00, 8.23343241e-01, 3.89293329e-01,
-1.09571764e-01],
[ 9.99741684e-01, 8.60251305e-01, 1.88082949e-01,
6.52864291e-01],
[ 1.35513821e+00, -4.12480819e-01, -4.92068858e-01,
6.43195491e-01],
[-5.50565262e-01, -1.47150461e+00, 2.93728037e-01,
-8.23140470e-02],
[-2.80141174e+00, 1.40228806e+00, 8.41263094e-01,
-1.44889914e-01],
[-9.63349112e-02, 1.99735289e-01, 1.17125418e-02,
2.11370813e-01],
[-2.16903379e-01, -9.70124183e-01, 6.24870938e-01,
-2.20847793e-01],
[-2.10858541e+00, 1.42484670e+00, 1.04774671e-01,
1.31908831e-01],
[-2.07971417e+00, -6.11268624e-01, -1.38864998e-01,
1.84103743e-01],
[-6.29426664e-01, 3.21012967e-01, -2.40659234e-01,
-1.66651801e-01]])